
Chemical cross-linking mass spectrometry (CXMS) is an important tool for resolving protein structures and studying protein interactions. The technique uses a chemical cross-linking agent to link two amino acids in close enough spatial proximity to each other in a covalent bond. The two amino acid sites where cross-linking occurs are identified by mass spectrometry. The distance between the two cross-linking sites should not exceed the arm length of the cross-linker due to the limitation of the cross-linker arm length. This distance information will provide important constraints for protein structure resolution and interaction studies.
Advantages of CXMS
(1) Sample preparation. The cross-linked proteins can be obtained by conventional proteomic digestion, purification and mass spectrometry, which is easy to operate and not limited by molecular weight and sample purity.
(2) High throughput. Based on the characteristics of mass spectrometry, the structure and interaction of multiple proteins can be analyzed simultaneously in high throughput.
(3) High sensitivity. Biological reactions are transient. Cross-linking agents can covalently link specific amino acids in transient interactions and poorly bound reactions, revealing transient or weak protein interactions.
(4) Clear mode of action: CXMS can determine whether proteins are interacting directly or indirectly, and in combination with crystal structure information can clarify the interaction interface information.
(5) Wide range of applications. In addition to cross-linking at the protein molecular level, some cross-linking agents can also enter the cell to cross-link the physiological state of the interaction at the cellular level.
Learn more: split ubiquitin assay
Workflow of CXMS
The workflow of a typical chemical cross-linking mass spectrometry technique consists of the following steps: cross-linker selection, cross-linking reaction, enzymatic digestion, cross-linked peptide enrichment, and liquid chromatography-mass spectrometry coupling analysis.
 General CLMS workflow (O'Reilly et al., 2018)
General CLMS workflow (O'Reilly et al., 2018)
Protein cross-linking reaction is one of the key steps in CXMS analysis, which directly affects the success of experimental results. When studying the interaction interface of subunits of simple proteins or protein complexes, a suitable cross-linking agent should be selected in advance according to the structure of the target protein and the structural domain of interest. Cross-linking is performed under conditions that do not affect the normal conformation of the protein and have a high reactivity of the cross-linker. Match the cross-linking agent with the protein and the buffer system by determining the reaction ratio between the cross-linking agent and the target protein, the reaction acidity and reaction time, etc. Avoid disrupting the natural structure of the protein and causing over or under cross-linking reaction with false information of under or pseudo cross-linking. The length of the intermediate arm of the cross-linker is also an important influencing factor when studying unknown protein interactions. In addition, cross-linking can be performed in vivo and in vitro. To capture the structure of a protein in its native state, the cross-linking reaction needs to be performed intracellularly.

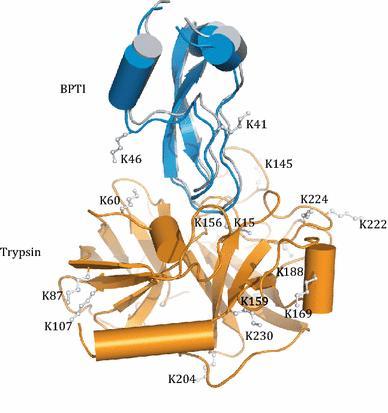
Visualizing the Ensemble Structures of Protein Complexes Using Chemical Cross-Linking Coupled with Mass Spectrometry (Gong et al., 2015)
After the cross-linking reaction of proteins, the cross-linking product enzymes need to be cut into peptide segments for mass spectrometry detection. The conventional trpsin is usually used to cleave the C-terminus of arginine (Arg) and lysine (Lys) for the enzymatic cleavage reaction. In addition to the use of trypsin, the addition of four proteases aspartate (Asp)-N, glutamate (Glu)-C, Lys-C and Lys-N for multi-enzyme cleavage can increase the number of cross-linked sites identified.
The specific enrichment and separation of cross-linked peptides can improve the signal intensity of cross-linked peptides and reduce the complexity of the sample, improving the efficiency of cross-linked peptide identification. Depending on the time of enrichment, there are pre-digestion protein enrichment and post-digestion peptide enrichment.
Pre-digestion protein enrichment refers to the purification or targeted enrichment of organelles or target proteins of interest. Combining cross-linking with affinity purification, capturing the target protein complex after the cross-linking reaction by affinity purification or capturing the target protein complex before the cross-linking reaction can effectively reduce the complexity of cross-linked peptide identification.
Cross-linked proteins can also be separated by SDS-PAGE in-gel with enzymatic digestion. During mass spectrometry data acquisition, the identification of cross-linked peptides can be improved by setting secondary mass spectrometry conditions for acquisition and collecting only secondary mass spectrometry information for the higher valence ions.
Peptide enrichment after digestion refers to the separation of peptides that undergo cross-linking from non-cross-linked linear peptides, and the commonly used method is size exclusion chromatography (SEC). Using SEC analysis, cross-linked linear peptides are selected based on their higher molecular weight, thus reducing the majority of non-cross-linked linear peptides present in the cross-linked system. In addition, due to the increased charge number of the cross-linked peptides, the cross-linked peptides possess more positively charged basic amino acids than the linear peptides, which can be enriched and separated using strong cation exchange chromatography (SCX).
The digested peptides are entered into mass spectrometry for data acquisition. Whether the cross-linker is mass spectrometry breakable or not has a significant impact on the choice of mass spectrometry fragmentation method for cross-linked peptides.
Since the cross-linker of unbreakable cross-linked peptides does not break easily in mass spectrometry, the main goal of the fragmentation method for such cross-linked peptides is to fragment the peptide sufficiently for subsequent identification of the cross-linked peptide, generally choosing higher energy ollisional dissociation (HCD) fragmentation.
The fragmentation of a breakable cross-linked peptide requires a balance between two aspects. On the one hand, it is desirable to fragment the cross-linker to obtain the complete peptide ion peaks of both peptides to infer the mass of the cross-linked two peptides and thus reduce the complexity of identification. On the other hand, it is desirable to fragment the peptide sufficiently to obtain as many peptide fragment ions as possible to identify the peptide sequence. However, these two goals are often in conflict with each other. The high abundance of intact peptide ion peaks implies that peptide fragmentation is not sufficient, so that fewer peptide fragment ions are produced. In addition, the bond energy of the fragile bonds in the cross-linker is often smaller than that of the peptide bond, making the cross-linker easier to fragment at lower energies than the peptide, resulting in a single fragmentation that often does not yield sufficient intact peptide ion peaks and peptide fragment ion peaks at the same time.
All current fragmentation methods have certain shortcomings, but one of the most commonly used fragmentation methods is the step energy based stepped collision energy HCD (SCE-HCD). This method is more efficient in data acquisition and not limited by the instrumentation. It is able to scan both intact peptide ion peaks and peptide fragmentation ion peaks in the same secondary spectrogram.
CX-MS can be combined with protein 3D structure prediction software to determine the spatial conformation of proteins, to fix weak/transient interactions, to help distinguish between direct and indirect interactions, and to determine the interface of protein interactions.
References
- O'Reilly, F. J., & Rappsilber, J. (2018). Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nature structural & molecular biology, 25(11), 1000-1008.
- Gong, Z., Ding, Y. H., et al. (2015). Visualizing the ensemble structures of protein complexes using chemical cross-linking coupled with mass spectrometry. Biophysics reports, 1(3), 127-138.